FAG Oversiktsartikkel
Cellefritt tumor-DNA i blod, et nytt verktøy for diagnostikk og monitorering av kreftpasienter?
De siste tiårenes inntog av sensitive molekylære metoder, som massiv parallell sekvensering (MPS) og digital PCR (dPCR), har bidratt til en økende interesse for analysering av cellefritt tumor-DNA.
Last ned pdf:
Cellefritt tumor-DNA i blod.pdf
Allerede i 1948 ble det påvist cellefritt DNA (cfDNA) i varierende mengde i blod, men først flere tiår senere oppdaget man at cfDNA i blodplasma også kunne stamme fra solide svulster. Mengden av cellefritt tumor-DNA (ctDNA) er ofte svært liten og det er i tillegg fragmentert, noe som har vanskeliggjort deteksjon. De siste tiårenes inntog av sensitive molekylære metoder, som massiv parallell sekvensering (MPS) og digital PCR (dPCR), har ført til en økende interesse for analysering av ctDNA. Vi gir her en oversikt over kunnskap innen feltet og diskuterer mulighetene for at måling av ctDNA i perifert blod kan benyttes som en klinisk-diagnostisk test hos pasienter med solide svulster.
Utvikling av kreft skyldes en rekke faktorer. Man antar at utgangspunktet er at en celle får vekstfordeler og er opphav til mange celler som etter hvert danner en svulst. Noen av disse cellene kan få genetiske endringer som gir dem evne til å migrere og invadere omkringliggende vev. Dette gjør at en svulst blir malign, men i tillegg kan kreftcellene få egenskaper som gjør at de kan trenge inn i blod- eller lymfebaner og metastasere til andre steder i kroppen. Cellegift og stråling, i tillegg til kirurgi, er fortsatt primærbehandling for de aller fleste kreftformer, men veien går mot en mer persontilpasset kreftbehandling med medikamenter rettet mot signalveier som har betydning for kreftcellenes overlevelse og vekst. Målrettet behandling er kostbar og kan være forbundet med betydelige bivirkninger, så når dette tilbys er det viktig å ha gode verktøy for seleksjon av pasienter, for deretter å måle om behandlingen er effektiv.
Materiale og metode
Artikkelen er basert på publikasjoner funnet ved søk i PubMed (søkeord: liquid biopsi, ctDNA, cfDNA, cancer), med siste søk i mai 2015. Vi har også inkludert egne erfaringer.
Cellefritt DNA (cfDNA)
Cellefritt DNA er segmenter av DNA som befinner seg utenfor celler. Man har i hovedsak studert cfDNA i blod, men det forekommer også i andre kroppsvæsker. Hovedvekten av cfDNA i blod stammer fra blodets egne celler, men også DNA fra andre celler entrer sirkulasjonen. Ved infeksjoner kan DNA fra patogener også være tilstede (1, 2). Opphavet til cfDNA antas i hovedsak å være celledød, både ved apoptose og nekrose, men det er også mulig at celler aktivt frigir DNA.
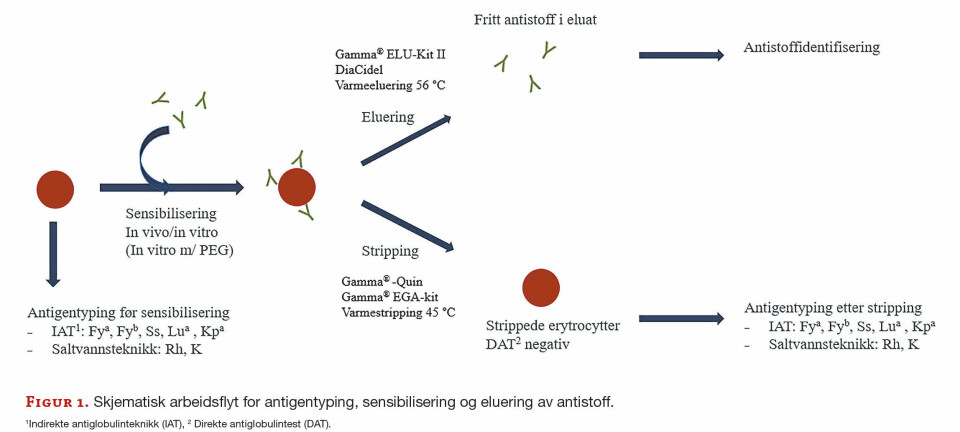
Ved nekrose sprekker cellene, cellerestene fordøyes av makrofager og det dannes DNA-fragmenter av varierende størrelser. Apoptose er en kontrollert prosess, styrt av cellens egne signaler. Cellen vil krympe, kromatinet kondenseres og kuttes til nukleosomale enheter, som består av 146 basepar (bp) DNA snurret rundt en histonkjerne. Ved apoptose får man DNA-fragmenter tilsvarende ett, to eller flere nukleosomer (figur 1A og 1B) (3).
Figur 1A

I cellekjernen er DNA bundet til proteiner og danner kromatin. Nukleosomet, den primære bestanddelen, består av en histonkjerne som DNA-heliksen er kveilet to ganger rundt, tilsvarende 146 bp, samt en liten "linkertråd" på mellom 15-100 bp. Histon H1 er med på å stabilisere og pakke nukleosomene. Det er i linker-regionen DNA er mest tilgjengelig og utsatt for degradering av nukleaser. Ved apoptose pakkes kromatinet, og endonukleaser kløyver DNA slik at det dannes DNA-fragmenter tilsvarende ett eller flere nukleosomer. Dette vil skape et mønster som kalles apoptotisk stige. Ved nekrose er ikke DNA like tett pakket slik at fragmenter av mer varierende lengder dannes.
Figur 1B
Konsentrasjon av cfDNA målt med Agilent High Sensitivity DNA Kit. X-aksen angir fragmentenes bp-størrelse og Y-aksen viser mengden cfDNA målt i forhold til øvre markør (10380 bp) og er oppgitt som fluoresensenhet (FU). En øvre og nedre (35 bp) markør ses i hvert elektroferogram.

I serum kan man tydelig se en apoptotisk stige med DNA-fragmenter tilsvarende ett, to og tre nukleosomer. Det er ca. 8-10 ganger høyere nivå av cfDNA i serum enn i EDTA-plasma, og dette stammer fra leukocytter som lyserer under koaguleringsprosessen.

En rekke tilstander, som betennelser, traumer, graviditet (føtalt DNA i mors blod), autoimmune sykdommer og fysisk belastning, fører til en økning av cfDNA. Vanligvis vil makrofager infiltrere vevet og fjerne de apoptotiske og nekrotiske cellerestene, men når systemet overbelastes, slippes cfDNA ut i sirkulasjonen (4). I blodbanen er cfDNA oftest bundet til proteiner, lipider og cellemembraner eller innkapslet i vesikler (5), og halveringstiden er kort (15 – 120 minutter) (6, 7).
Totalmengde cfDNA i blod hos kreftpasienter
Allerede i en studie fra 1977 ble det undersøkt om totalmengde cfDNA i serum var høyere hos kreftpasienter enn hos friske individer. Selv om det i snitt ble påvist et betydelig forhøyet nivå hos kreftpasienter, fant de også store forskjeller mellom de ulike kreftformene. Halvparten av pasientene hadde normalt nivå, men det kunne skyldes at pasientene allerede var behandlet med kirurgi eller kjemoterapi (8). Senere studier har støttet disse funnene, og generelt finner man økt mengde cfDNA hos kreftpasienter sammenliknet med friske, og nivået henger ofte sammen med tumormasse (9), men variasjonen er stor og ofte overlappende med benigne tilstander.
Flere har også undersøkt om fragmentstørrelsen til cfDNA kan skille mellom friske, benigne og maligne tilstander. I en studie ble det påvist en sammenheng både mellom økt nivå og økt fragmentering av cfDNA hos pasienter med kolorektalkreft, sammenliknet med friske individer. Tilsvarende så man en tendens til økt mengde og mindre intakt cfDNA ved metastatisk bryst- og kolorektalkreft, sammenlignet med friske og pasienter med lokalisert sykdom (10, 11). Andre studier har kommet til motsatt konklusjon og påvist økt andel lange fragmenter hos kreftpasienter, sammenliknet med friske. En av teoriene var at lange fragmenter skyldtes nekrotisk tumorvev (12 -14). Det foreligger også studier hvor slike sammenhenger ikke ble påvist (15).
Det er derfor fortsatt uavklart om fragmentering og totalmengde cfDNA er informativt. En stor utfordring er mangel på standardiserte metoder, noe som bør etableres for å kunne avklare om mengde og lengde av cfDNA kan benyttes som markører ved kreftsykdom.
Cellefritt tumor-DNA (ctDNA)
Ettersom en rekke benigne tilstander fører til økt cfDNA, er deteksjon av somatiske mutasjoner en metode for å påvise ctDNA.
Over tid akkumulerer kreftcellene en rekke mutasjoner, de fleste vil være «passasjermutasjoner» som ikke er involvert i kreftutviklingen, mens en liten andel vil være aktive pådrivere av sykdommen, såkalte «sjåførmutasjoner». Når man leter etter ctDNA bør man velge «sjåførmutasjoner», som har oppstått tidlig og finnes i alle cellene. Men man må være oppmerksomme på at det kan finnes subkloner med andre mutasjoner som kan dominere senere i sykdommen.
Flere store forskningssamarbeid, for eksempel The Cancer Genome Atlas (TCGA) og International Cancer Genome Consortium (ICGC) har kartlagt og offentliggjort mutasjonsspektrene for en rekke kreftformer. Denne informasjonen kan brukes til valg av markører. Ved noen typer kreft, som kolorektalkreft, oppstår mutasjoner i hotspotregioner. Det er da tilstrekkelig å lete etter mutasjoner i et fåtall gener med tanke på behandling. Ved andre typer kreft, som for eksempel brystkreft, er bildet mer heterogent, og det er bare noen få gener som er oftere mutert enn andre. Dermed er det ikke nødvendigvis tilstrekkelig å etablere en screeningtest basert på noen få gener for hele pasientgruppen. I tillegg er noen kreftsykdommer mer dominert av kopitallsforandringer enn andre, noe som krever en egen tilnærming (16).
Svulster er i utgangspunktet klonale, det vil si at de utgår fra én celle, men når svulsten vokser kan det oppstå subkloner av celler med andre mutasjoner. Både under kreftutvikling og ved behandling, er det en risiko for at det oppstår subkloner som gjør kreftceller motstandsdyktige og/eller gir celler evne til å spre seg. Derfor vil én enkelt biopsi av en svulst ikke nødvendigvis være representativ for alle subkloner. I tillegg er det ikke mulig å ta biopsier fra alle metastaser, slik at muligheten for å kartlegge heterogenitet er begrenset. En løsning kan være deteksjon av ctDNA i blod. Dersom tumor-DNA fra alle kloner kommer ut i blodbanen, kan analyse av ctDNA i blodprøve gi et bedre helhetsinntrykk av hvilke mutasjoner som er tilstede (figur 2). En slik tilnærming vil både spare pasienten for smertefulle inngrep og helsevesenet for avansert prøvetakning og kostbar radiologi. Samtidig kan det gi legene mulighet til tidlig endring av behandlingsregime.
Figur 2

Metoder for deteksjon av ctDNA i blod
Detaljer om prøvetaking og metodebeskrivelser for massiv parallell sekvensering (MPS) og digital PCR (dPCR) er presentert i egne faktabokser.
Plasma foretrekkes fremfor serum for isolasjon av cfDNA, fordi leukocytter lyseres og frigjør DNA under koagulasjonsprosessen (figur 1B) (17). Siden mengden ctDNA ofte er lav i forhold til mengden fritt normalt DNA, må man benytte sensitive teknikker, men valg av metode avhenger også av hvilken krefttype som skal undersøkes. MPS er egnet til å screene et stort antall gener for mutasjoner, mens dPCR kun analyserer et fåtall gener samtidig. En forskningsgruppe benyttet dråpebasert dPCR (ddPCR) og analyserte de syv vanligste KRAS-mutasjonene i plasma-DNA fra 50 pasienter med metastatisk kolorektalkreft. Resultatene viste god korrelasjon, både når mutasjonstestene ble analysert hver for seg og samlet i en multipleksanalyse, men multiplekstesten hadde noe lavere sensitivitet (18). En ulempe med dPCR er at man på forhånd må vite hvilke mutasjoner man vil detektere, eventuelt må man bestemme seg for å detektere bare hyppig forekommende forandringer. Men dPCR er enkel å utføre og har god sensitivitet, med mulighet til å detektere < 0,01 % mutert DNA.
MPS er per i dag ikke like sensitivt som dPCR, men fordelen er at man kan sekvensere opp til flere tusen genregioner parallelt, og screene mange prøver samtidig, selv om pasientgruppen er heterogen. I en studie (19) ble det valgt ut 500 gener for målrettet sekvensering av cfDNA fra pasienter med ikke-småcellet lungekreft. Analysen ble først utført på primærtumor for identifisering av hvilke mutasjoner som var til stede hos hver av pasientene, deretter ble analysen utført på cfDNA for deteksjon av ctDNA. I snitt ble seks mutasjoner detektert i tumor hos hver pasient. Deteksjonsgrensen gikk helt ned mot 0,02 % i plasma når man visste hvilke mutasjoner det skulle letes etter. Ved analyse av ctDNA uten å ha kjennskap til mutasjonsspekteret på forhånd var deteksjonsgrensen på 0,4 % (19).
Monitorering av sykdomsforløp ved hjelp av ctDNA
For mange kreftformer bruker man, i tillegg til klinisk undersøkelse, serumanalyser for kreftantigener som CEA, CA125, CA 15-3 og PSA for monitorering. Disse analysene har i en del tilfeller manglende spesifisitet og sensitivitet. Når det gjelder radiologi kan både tumorstørrelse og påvisning av metastaser underveis i behandlingen være utfordrende.
Ved enkelte leukemier har man i mer enn ti år rutinemessig målt behandlingsrespons i blod eller benmargsprøver ved å påvise tumorspesifikke endringer i hvite blodlegemer. Svært sensitive metoder har gjort det mulig å finne én leukemisk celle blant 100 000 normale celler, såkalt minimal restsykdom (MRD). Endringer i MRD-verdiene sier noe om respons på behandling og brukes for å velge behandlingsregimer underveis (20, 21). For solide svulster har det ikke vært like enkelt å detektere MRD. Det har vært gjort mye arbeid basert på deteksjon av sirkulerende tumorceller i blod (CTC) og disseminerte tumorceller i benmarg (DTC) fra solide svulster. Men frekvensen av slike sirkulerende kreftceller er relativt lav (gjerne 1 av 10 000 000 celler). Og i tillegg kan falskt negativt resultat være en utfordring, fordi cellene identifiseres ved hjelp av epitelspesifikke proteiner, som kan være underuttrykt.
I en studie ble pasienter under behandling for metastatisk brystkreft, monitorert med måling av ctDNA, CTC og CA15-3, i tillegg til standard radiologiundersøkelse. Formålet var å undersøke hvor informativ ctDNA var når det gjaldt respons på terapi, sammenlignet med de andre markørene. Resultatene viste at ctDNA korrelerte best (89 %) med de radiologiske funnene og at ctDNA varslet tilbakefall to til ni måneder tidligere enn radiologi hos 53 % av pasientene (22). I en annen studie hvor man analyserte ctDNA i plasma hos pasienter med kolorektalkreft, fant man ctDNA i blod ti måneder tidligere enn sykdomsprogresjon påvist ved radiologi (23). Dette indikerer at man kan bruke deteksjon av ctDNA for å endre behandling. Det gir større mulighet til å forhindre metastatisk sykdom.
En annen studie har vist at mengde ctDNA kan variere, selv hos pasienter med fremskreden sykdom, og at nivået ikke nødvendigvis reflekterer grad av sykdom. Ved hjelp av detaljundersøkelser av prøver fra én brystkreftpasient med store mengder sirkulerende tumorceller, fant man kun 3 % ctDNA i plasma. I en tidligere studie hadde samme forskningsgruppe, hos en andel av pasienter med metastastisk kolorektal- eller brystkreft, sett en sammenheng mellom økt antall CTC, økte fragmentlengder og økt nivå av ctDNA (24, 25).
De fleste studiene hittil har fokusert på påvisning av ctDNA hos pasienter med metastatisk sykdom, ettersom konsentrasjonene er høyest hos denne gruppen. Noen få studier har også vist at ctDNA er detekterbart i tidlig stadium. En studie som omfattet 14 krefttyper der ctDNA ble undersøkt hos pasienter både i tidlig og sen fase av sykdom, viste at 49 % av pasientene i tidlig stadium hadde detekterbart ctDNA, kontra 82 % av pasienter med metastatisk sykdom. De påviste også store forskjeller i mengde ctDNA hos pasienter med ulike kreftformer. Også innad i samme kreftsykdom kunne mengde ctDNA variere betydelig (26). I en liten, prospektiv studie ble pasienter med brystkreft i tidlig stadium analysert for ctDNA før og etter operasjon. Selv om nivået var lavt hos de fleste før operasjon, kunne man fortsatt påvise små konsentrasjoner av ctDNA hos 50 % av pasientene hvor det var tatt postoperativ blodprøve (27).
Dersom ctDNA skal brukes for monitorering, er standardisert prøvetakning og prosessering viktig, slik at det kvantitative nivået av ctDNA er sammenlignbart for hver enkelt pasient gjennom hele forløpet. Selv om det letes etter spesifikke molekylære tumormarkører med sensitive metoder, viser studier at ctDNA ikke er påvisbart hos enkelte pasienter, for eksempel ved progressiv metastatisk sykdom (25). Dette kan skyldes at ctDNA faktisk ikke er tilstede i blod, men man må også vurdere om det kan være metodiske årsaker. Mens man i mange studier av ctDNA har benyttet fragmentlengder på rundt ett nukleosom, har andre påvist at ctDNA kan detekteres ved analyse av kortere fragmentlengder. Dette kan forklare hvorfor enkelte rapporterer om høyere andel ctDNA i kolorektalkreft, sammenlignet med andre studier, når analysene var tilpasset fragmentlengder mindre enn 100 bp (28–30).
For oppfølgning av enkeltpasienter er det i tillegg viktig å velge best egnet metode for måling av ctDNA gjennom hele sykdomsforløpet. Bare små endringer i fragmentlengder kan gi stor innvirkning på kvantitative målinger. Siden utviklingen av metoder for dPCR og MPS skjer raskt, er det viktig å ta dette med i betraktning hvis man endrer metoder underveis.
Deteksjon av ctDNA i blod; utfordringer og muligheter
En utfordring ved deteksjon av ctDNA er at de mest sensitive metodene, slik som dPCR, krever at man vet hva man leter etter. For enkelte tumortyper med få høyfrekvente mutasjoner forutsettes det at man kjenner mutasjonsprofilen for den enkelte pasient, for deretter å kunne skreddersy tester til oppfølgning. I forskningsstudier er dette viktig for å kunne kartlegge betydningen av slik monitorering, men som rutinediagnostikk kan det være litt for tid- og ressurskrevende. Utviklingen innen MPS går raskt, og muligheten for å gjøre sekvensering av mange selekterte gener med lite startmateriale og høy sensitivitet, er snart innen rekkevidde. Det forventes også at både pris og tidsbruk vil falle betraktelig. Det muliggjør design av målrettede sekvenseringsanalyser for hver tumorform, for både kartlegging av DNA-forandringer i primærtumor, og deteksjon av ctDNA i oppfølgningsprøver. En fordel med å undersøke genpaneler er at man kan detektere forandringer i ctDNA som ikke var tilstede i primærtumoren.
Digital PCR (dPCR)

Ved dPCR benyttes spesifikke primere i kombinasjon med fluoriserende fargestoffer eller prober som binder seg til DNA, slik at dannelsen av PCR-produkt kan måles. Det finnes ulike instrumenter på markedet i dag for dPCR, for eksempel dråpebaserte metoder (ddPCR) og metoder med mikrokamre. Dråpebaserte systemer kan generere mange millioner dråper fra ett reaksjonsvolum, mens mikrokammer-metoder ofte genererer noe færre antall reaksjoner per prøve. Hver dråpe/kammer er av picoliterstørrelse hvor alle PCR-reagenser, prober, primere og templat er tilstede. Det som er viktig ved dPCR er å optimalisere mengde templat slik at man i størst mulig grad oppnår ett DNA-fragment i hver dråpe/kammer.
Illustrasjonen viser et eksempel på en ddPCR hvor fem analyser utføres i samme eksperiment. Systemet kan kun avlese fluorescens i to bølgelengdeområder, men man kan likevel kombinere flere tester samtidig (multipleks), ved å justere mengde probe (signalstyrke) og bruke prober med to fluoroforer for én analyse. Etter PCR telles og skilles dråpene fra hverandre utfra størrelse og fluorescens ved hjelp av væskestrøm. Hver prikk i diagrammet tilsvarer én dråpe. Diagrammet viser signalstyrken til FAM-probene langs X-aksen, MAX-probene langs Y-aksen og testen med både FAM- og MAX-probe i midten. Nede til venstre i diagrammet er bakgrunnsfluorescens fra dråper uten PCR-produkt. Positive kontroller benyttes til å identifisere hvor signalet fra de ulike testene skal ligge. Dråper utenfor de avgrensede områdene kan være dråper hvor det har kommet mer enn ett templat eller støy fra ødelagte dråper. Normalt vil dette ikke påvirke resultatet siden man har flere millioner dråper.
En annen utfordring ved deteksjon av ctDNA er presis kvantitering. Ettersom en økning av ctDNA ser ut til å være informativ, må protokollene være standardiserte slik at variasjonen mellom prøver fra ulike tidspunkt er minimale. I en EU-finansiert studie vi deltar i, arbeides det med å utvikle aprotokoller for prøvetakning, prosessering og ekstraksjon som vil kunne ISO-sertifiseres. Det er i tillegg av betydning hvor stort blodvolum man ekstraherer DNA fra, og hvor mye DNA man tar videre i analysen. Har man mye cfDNA (for eksempel ved infeksjon) er totalmengden cfDNA høy og man må undersøke mer DNA for å finne ctDNA. Mange oppgir derfor mengde ctDNA per ml plasma som standard. Ved design av assay for dPCR, er det også av betydning hvor lang fragmentlengde man undersøker. Ved negative prøver bør man alltid vurdere om det kan være metodiske årsaker til at man ikke klarer påvise ctDNA.
Konklusjon
Deteksjon av ctDNA er et felt som er i rask utvikling, både kunnskapsmessig og teknologisk. Kombinasjon av økt fokus på standardisering av prøvetakning og målemetoder, og at flere kliniske studier velger å inkludere ctDNA-målinger, vil på sikt gi svar på om ctDNA-analyser skal inn som rutine for oppfølgning av kreftpasienter. Denne metoden kan bli et viktig bidrag til bedre monitorering av behandlingsrespons og sykdomsprogresjon hos kreftpasienter.
Takk
Alle tre forfattere mottar støtte fra Helseregion Helse Sør-Øst for forskning og metodeutvikling. Takk også til Den Norske Kreftforening og til K. G. Jebsen forskningsstiftelse for økonomisk støtte.
Interessekonflikter
Ingen av medforfatterne har interessekonflikter å melde.