FAG Originalartikkel
DNA-ekstraksjon: Mengde er ikke synonymt med mangfold
Hovedbudskap:Før enhver molekylærgenetisk analyse bør metode for ekstraksjon av nukleinsyrer evalueres.Høyt utbytte av bakterielt DNA er ikke ensbetydende med stort bakterielt mangfold/diversitet.
DNA_ekstraksjon_Mengde er ikke synonymt med mangfold.pdf(250 KB)
Introduksjon
Alle molekylærgenetiske analyser starter med frigjøring eller ekstraksjon av nukleinsyrer (DNA og/eller RNA) fra celler. Når nukleinsyrer er tilgjengeliggjort, kan en rekke nedstrømsanalyser utføres for å studere ulike parametere i DNA- og RNA-sekvenser. Avhengig av hva slags analyse som skal utføres, stilles det ulike krav til både renhet, integritet, konsentrasjon og mangfold av nukleinsyrer. Det er derfor viktig at ekstraksjonsprosessen vies nok tid og oppmerksomhet når nye metoder skal etableres, og at denne prosessen evalueres og optimaliseres for hver enkelt analyse og hvert enkelt prøvemateriale.
De senere årene har tarmens bakteriesammensetning, mikrobiota, fått mye oppmerksomhet. Mikrobiotaen ser ut til å ha betydning for utvikling eller persistens av ulike sykdommer og tilstander, som for eksempel matintoleranse, diabetes, kronisk inflammatoriske sykdommer og kolorektal kreft (1-3). Feces er et hyppig benyttet prøvemateriale fordi det er lett tilgjengelig uten invasiv prøvetaking. Tykktarmen er det mest bakterierike organet i menneskekroppen med 1012 bakterier per gram feces (4). Bakterier representerer 25-54 prosent av biomassen i feces, og humant DNA kun 0,1 promille av total DNA-mengde (5). Tilstedeværelse av humant DNA i feces kan blant annet benyttes som markør for økt celledeling og kolorektal kreft (6-8). Dersom man skal analysere både humant og bakterielt DNA fra samme prøveeluat, må man sørge for at metoden man benytter gir tilfredsstillende utbytte av begge varianter.
Bakteriene i feces domineres av bakterierekkene Firmicutes og Bacteroidetes (4). Bakterier i de to rekkene har ulik oppbygning av celleveggen. Firmicutes er klassifisert som gram-positiv og Bacteroidetes som gram-negativ. Gram-positive bakterier er hardføre og robuste sammenliknet med gram-negative bakterier. De er derfor vanskeligere å lysere i laboratoriet for å få frigjort DNA. Når man skal studere bakteriell diversitet i en prøve, må ekstraksjonsmetoden optimaliseres for jevn frigjøring av DNA fra alle mikrober i prøven, slik at man får et representativt bilde av bakteriesammensetningen. Det skilles ofte mellom alfadiversitet og betadiversitet ved studier av bakteriesammensetninger. Alfadiversitet tar høyde for både artsrikdom og relative mengder i et avgrenset miljø, gjerne i en biologisk prøve, mens betadiversitet betegner forskjellen mellom ulike miljøer, eksempelvis mellom biologiske prøver (9). Flere studier har vist at ulike metoder for DNA-ekstraksjon resulterer i forskjellig utbytte og diversitet, noe som i stor grad påvirker tolkningen av resultatene (10-12). Feces inneholder også en rekke substanser som kan hemme nedstrømsapplikasjoner. Det er derfor viktig at det benyttes gode renseprosesser ved nukleinsyreekstraksjon fra feces, slik at hemmende substanser fjernes.
Som et ledd i en kvalitetssikringsprosess av metoder til en større studie om kolorektal kreft, har vi ekstrahert DNA fra feces fra 22 friske, frivillige personer med fire ulike DNA ekstraksjonskit og totalt seks ulike protokoller. Målet med denne studien var å identifisere den beste DNA ekstraksjonsmetoden for å studere bakteriell diversitet og humant DNA i feces-prøver.
Materiale og metode
22 friske personer ansatt ved Akershus Universitetssykehus, Lørenskog, ble rekruttert til kvalitetssikringsstudien. Fecesprøvene ble samlet etter spesifikke skriftlige instruksjoner som alle frivillige fikk tildelt. Alle deltakere signerte et samtykkeskjema. Fordi formålet med studien var kvalitetssikring av metode, var godkjenning fra Regional Etisk Komité (REK) ikke påkrevet. Omtrent en teskje feces ble samlet fra tre ulike områder av materialet, og umiddelbart overført til samme sterile prøvetakingsrør med 4 ml RNAlater RNA Stabilization Reagent (ThermoFisher Scientific, Waltham, MA, USA). Prøvene ble oppbevart i romtemperatur eller kjøleskap i 18-24 timer, etter anbefalinger fra produsenten. Etter ankomst til laboratoriet ble prøvene anonymisert, homogenisert, allikvotert og fryst ved -80°C.
DNA-ekstraksjon
Valg av DNA ekstraksjonskit til utprøving ble foretatt på bakgrunn av litteraturstudier (11, 13, 14) og tilgjengelige instrumenter i vårt laboratorium. Et mekanisk lyseringstrinn ble innført der dette ikke var en del av protokollen. Valg av kuler til mekanisk lysering ble gjort på grunnlag av tidligere erfaringer (15). Innføring av et ekstra enzymatisk lyseringstrinn for to av protokollene ble foretatt etter anbefaling fra andre studier (16). Hver fecesprøve (n = 22) ble ekstrahert med seks ulike DNA ekstraksjonsprotokoller.
QIAamp® DNA Stool Mini Kit (Q)
Ekstraksjon med QIAamp® DNA Stool Mini Kit (Qiagen) ble utført med protokoll for patogen deteksjon etter produsentens anbefalinger (200 μl prøvemateriale). Et mekanisk lyseringstrinn ble lagt til etter trinn 3 i protokollen. Materiale i lyseringsbuffer ble overført til et Fast Prep “Soil” Lysing matrix E-rør (LME-rør) (MP Biomedicals, Santa Ana, USA) med tre typer kuler: 1,4 mm keramiske kuler, 0,1 mm silikakuler og en glasskule på 4 mm. LME-røret ble ristet ved 6 m/s i 3 x 60 sekunder i FastPrep®-24 (MP Biomedicals) med 60 sekunders pause mellom hver runde. Røret ble så sentrifugert ved 13 000 g i 1 minutt og cirka 1000 μl supernatant ble overført til et rør med InhibitEX fra samme kit. Kitets instruksjoner ble fulgt etter dette trinnet. Elueringsvolum var 100 μl (tabell 1).
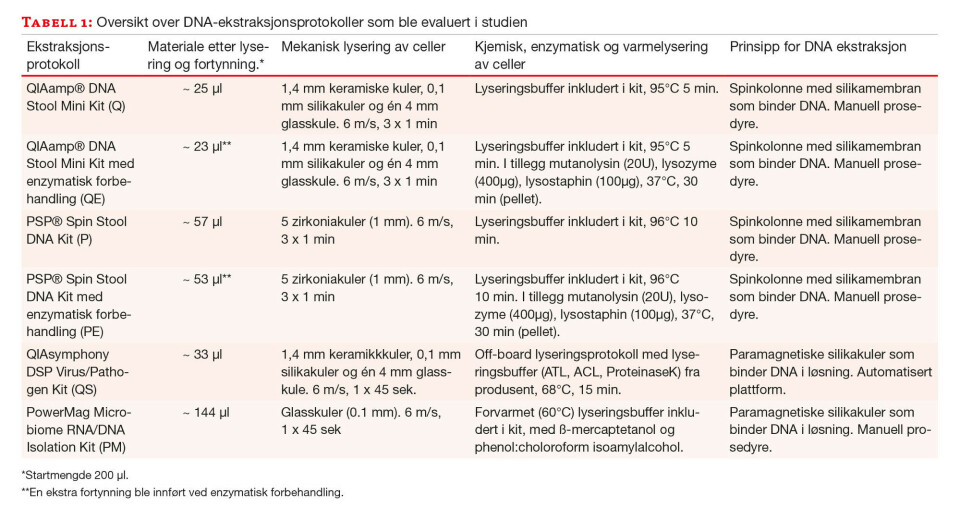
QIAamp® DNA Stool Mini Kit med enzymatisk forbehandling (QE)
Tilsvarende protokoll ble utført med inklusjon av et ekstra enzymatisk lyseringstrinn. Etter mekanisk lysering i FastPrep®-24 ble røret forsiktig sentrifugert ved 600 rpm for å pelletere kuler og store fecespartikler. Supernatanten (900 μl) ble overført til et nytt rør (rør 1) som ble sentrifugert ved 13 000 g i 1 min for å pelletere intakte bakterieceller. Supernatanten (800 μl) ble fjernet fra rør 1 og overført til et nytt rør (rør 2), mens pelleten i rør 1 ble enzymbehandlet som beskrevet i tabell 1. Materialet i rør 1 (160 μl) og rør 2 (800 μl) ble overført til et felles rør med InhibitEX fra samme kit og kitets instruksjoner ble så fulgt. Elueringsvolum var 100 μl (tabell 1).
PSP® Spin Stool DNA Kit (P)
Ekstraksjon med PSP® Spin Stool DNA Kit (Stratec Molecular Gmbh, Berlin, Tyskland) ble utført med protokoll for anrikning av bakterielt DNA etter produsentenes anbefalinger (200 μl prøvemateriale ble benyttet). Avvikende fra protokollen var utførelse av mekanisk lysering med FastPrep-24 (MP Biomedicals) ved 6 m/s i 3 x 1 minutt, fremfor vorteksing i 2 minutter som anbefalt av produsenten. Elueringsvolum var 100 μl (tabell 1).
PSP® Spin Stool DNA Kit og enzymatisk forbehandling (PE)
Tilsvarende protokoll ble utført med inklusjon av et ekstra enzymatisk lyseringstrinn. Etter mekanisk lysering i FastPrep®-24 ble røret forsiktig sentrifugert ved 600 rpm for å pelletere kuler og store fecespartikler. Supernatanten (900 μl) ble overført til et nytt rør (rør 1) som ble sentrifugert ved 13 000 g i 1 min for å pelletere intakte bakterieceller. Supernatanten (800 μl) ble fjernet fra rør 1 og overført til et nytt rør (rør 2), mens pelleten i rør 1 ble enzymbehandlet som beskrevet i tabell 1. Materiale fra rør 1 (160 μl) og rør 2 (800 μl) ble overført til et felles InviAdsorb rør og kitets instruksjoner ble så fulgt. Elueringsvolum var 100 μl (tabell 1).
QIAsymphony DSP Virus/Pathogen Kit (QS)
Prøvemateriale (200 μl) ble overført til et LME-rør med 1 ml PBS (pH 8) før mekanisk lysering på FastPrep®-24 i 45 sekunder ved 6 m/s. Rørene ble sentrifugert 1 minutt ved 600 rpm før 200 µl supernatant ble behandlet med Qiagens Pathogen complex off-board lysis protocol (tabell 1). Nukleinsyreekstraksjon ble deretter utført med QIAsymphony ekstraksjonsinstrument og QIAsymphony DSP Virus/Pathogen protokoll, complex 200_OBL-V1. Elueringsvolum var 100 μl (tabell 1).
PowerMag Microbiome RNA/DNA Isolation Kit (PM)
Prøvemateriale (200 μl) ble ekstrahert med PowerMag Microbiome RNA/DNA Isolation Kit (MoBio Laboratories. Inc., Carlsbad, CA, USA), etter protokoll fra produsenten, men tilpasset en manuell prosedyre. Avvikende fra protokollen var en mekanisk lysering med FastPrep®-24 (MP Biomedicals) ved 6 m/s i 45 sekunder, i stedet for en «plate well shaker» (MP Biomedicals) som var anbefalt av produsenten. Elueringsvolum var 100 μl.
Kvantifisering av bakterielt DNA
Real-time PCR rettet mot genet som koder for 16S ribosomalt RNA (16S rRNA) ble utført for å vurdere utbytte av bakterielt DNA. Kvantifisering ble utført ved hjelp av en standardkurve etablert med DNA fra bakterien Escherichia coli (E. coli Non-methylated Genomic DNA 5 µg/20µl (Zymo Research Corp, Irvine, CA, USA)). Alle prøver ble analysert i duplikater og fortynnet 1:100 slik at de lå innenfor standardkurven. Primerne som ble benyttet (17) er oppgitt i tabell 2. PCR-reaksjonen besto av 12,5 µl SYBR Premix EX Taq (Takara Bio, USA), 2 µl av hver av primerne BAC-338 og BAC-805 (Eurogentec, 0.4 µM sluttkonsentrasjon), 6 µl H2O (5Prime GmbH, Hilden, Tyskland), 0,5 µl ROX II reference dye (inkludert i SYBR Premix E EX Taq kit), og 2 µl templat i et sluttvolum på 25 µl. Rotor-Gene Q real time PCR instrument (Qiagen) ble benyttet med følgende innstillinger: 95 °C i 10 sekunder etterfulgt av 30 sykler med 95 °C i 3 sekunder og 60 °C i 25 sekunder.
Kvantifisering av humant DNA
Humant DNA-utbytte ble målt med et kommersielt kit (Quantifiler Human DNA Quantification Kit (ThermoFisher Scientific)) som detekterer genet for humant telomerase revers transkriptase (hTERT). Kvantifisering ble utført med en standardkurve som besto av syv trefolds fortynninger av hTERT (fra samme kit) analysert i triplikater. Prøvene ble analysert ufortynnet i duplikater med alle forhold som anbefalt fra produsenten. AB 7900 HT real time PCR instrument (ThermoFisher Scientific) ble benyttet til amplifisering.
Data for både bakterielt og humant DNA ble analysert med Shapiro-Wilks normalfordelingstest, og resultatene ble sammenliknet med ANOVA og Wilcoxon rangeringstest for parvise data. Ettersom det ble utført multiple analyser, ble p-verdiene Bonferroni-korrigert. Alle statistiske analyser ble utført i Analyse-it (Analyse-it Software, Ltd., Leeds, UK).
Bakteriell diversitet
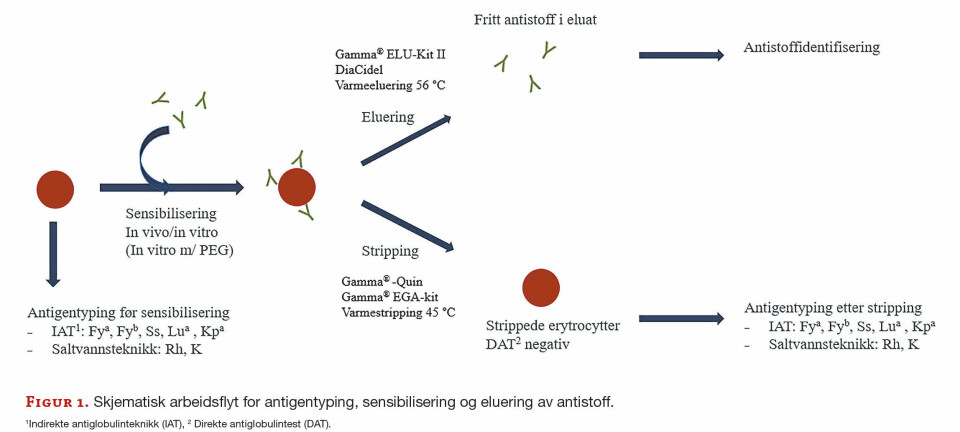
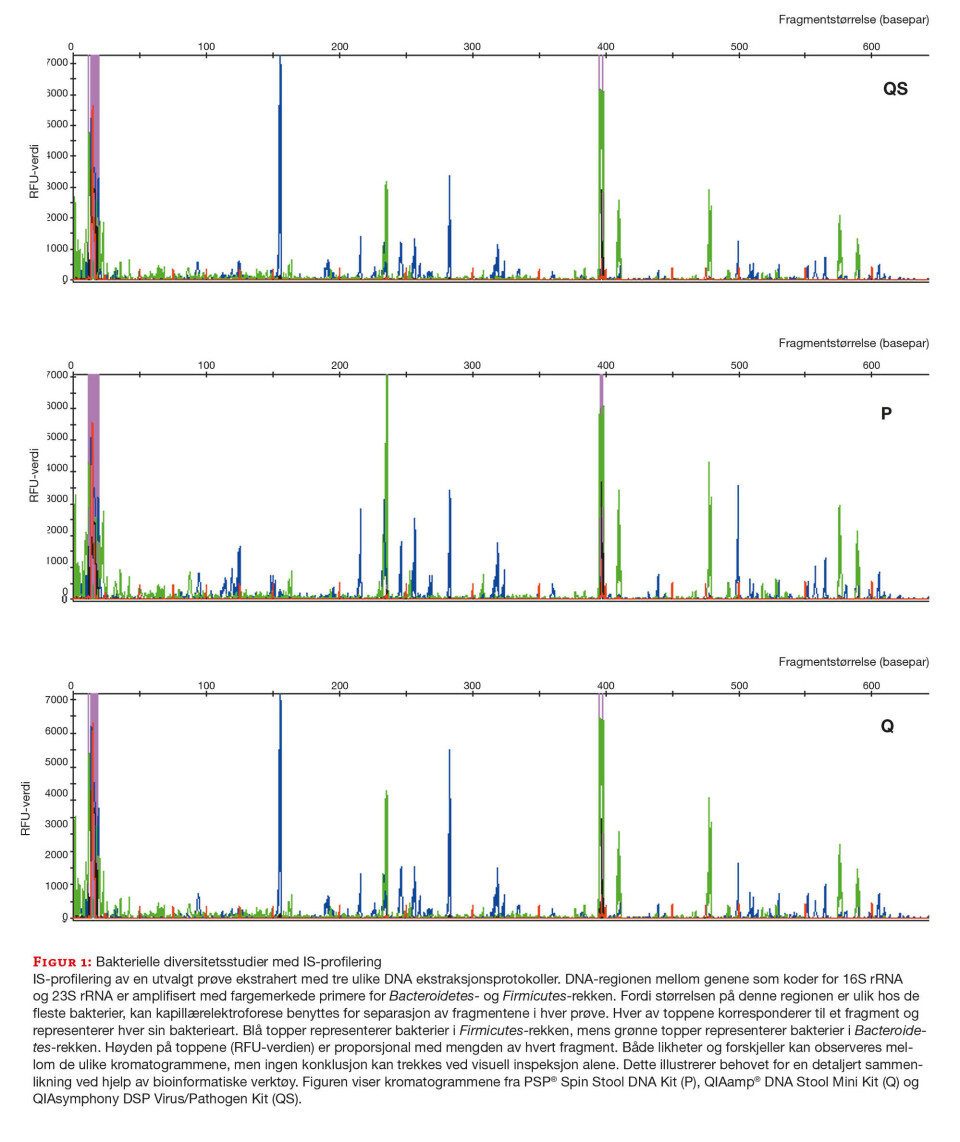
Bakteriell diversitet ble undersøkt ved IS-profilering, et begrep innført av Budding og medarbeidere i 2012 (18). DNA-regionen mellom genene som koder for 16S rRNA og 23S rRNA (Intergenisk Spacer, også kalt Intertranscribed spacer) ble amplifisert i hver prøve, og alle IS-fragmenter ble så separert med kapillærelektroforese (figur 1).
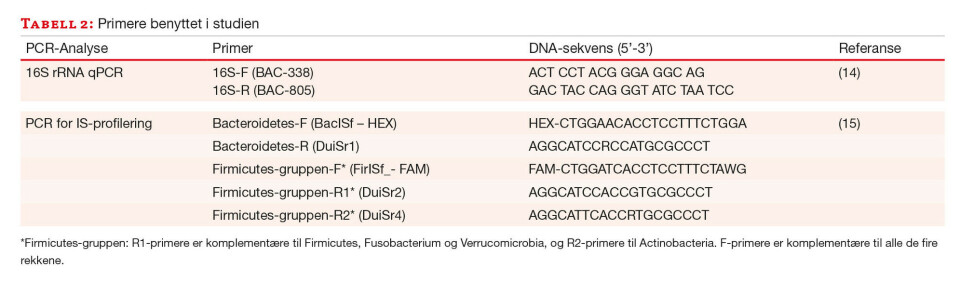
En multiplex PCR med to ulike primersett spesifikke for henholdsvis Bacteroidetes (HEX-merket) og Firmicutes, Actinobacteria, Fusobacterium og Verrucomicrobia (FAM-merket) ble benyttet. PCR-reaksjonen besto av 1 µl av hver av primerne FirISf_(FAM), BacISf_(HEX), DuiSr1, DuiSr2 og DuiSr4 (sluttkonsentrasjon på 0,04 µM), 12,15 µl H2O, 2,5 µl Invitrogen 10x PCR buffer -MgCl, 1,75 µl 50mM MgCl2, 0,5 µl 10mM dNTP, 1 µl BSA (1 %), 0,1 µl Platinum Taq polymerase og 2 µl templat (DNA fortynnet 1:10) i et sluttvolum på 25 µl. Rotor-Gene Q real time PCR instrument (Qiagen) ble benyttet med følgende innstillinger: 95 °C i 2 minutter etterfulgt av 35 sykler med 94 °C i 30 sekunder, 56 °C i 45 sekunder, og 72 °C i 60 sekunder. Alle prøver ble analysert i to tekniske replikater i samme oppsett. Primere er oppgitt i tabell 2.
Kapillærelektroforese for separasjon av IS-fragmenter ble utført med ABI Prism 3130xl GeneticAnalyzer med et 50 cm kapillær og POP-7 polymer. 5 µl PCR-produkt, 0,1 µl Map Marker 1000 size standard (ROX) og 19,9 µl Hidi formamide (ThermoFisher Scientific) ble benyttet i reaksjonsmiksen. Modulen 1200LIZ size standard for 3130xl Genetic Analyzer ble benyttet med alle innstillinger som anbefalt fra leverandør.
Dataprosessering av fragmentlengdeprofiler fra kapillærelektroforese ble analysert med BioNumerics software versjon 7.1 (Applied Maths, Sint-Martens-Latem, Belgia). Filter for fargeblødning mellom kanaler ble satt til 7000 punkter. Grensen for å detektere en topp ble satt til 1 % av «OD range» og 2 % of «curve range». Fragmentene i markøren ble benyttet til normalisering og sammenstilling av prøveprofilene. For å fjerne potensielle primer-dimere, ble grensen for minste fragmentlengde satt til 60 basepar. Skyggebånd (feil fra polymerasen) ble fjernet dersom de var mindre enn 1,2 basepar unna og mindre enn 60 % av høyden til en topp. Alle innstillingene var som anbefalt fra leverandør. Negative kontroller ble benyttet til å vurdere kontaminering i reagenser eller uspesifikke topper.
Fragmentlengdeprofilene (antall fragmenter og mengde av hvert fragment) for hver prøve ble benyttet til beregning av bakteriell diversitet. Diversitet internt i hver prøve (alfadiversitet) ble beregnet i Bionumerics (Applied Maths) med Shannon diversitetsindeks som stiger proporsjonelt med diversiteten (19). Gjennomsnittlig Shannon diversitetsindeks for alle prøver fra hver ekstraksjonsmetode ble så beregnet, og Wilcoxon rangeringstest for parvise data ble benyttet til å vurdere forskjeller mellom hver ekstraksjonsprosedyre. Ettersom det ble utført flere sammenlikninger, ble alle p-verdier Bonferroni-korrigert. Diversitet mellom prøver isolert med samme ekstraksjonsprosedyre (betadiversitet) ble beregnet ved å konstruere et dendrogram i Bionumerics (Applied Maths). Pearsons korrelasjon ble benyttet på fragmentlengdeprofilene for å måle likhet mellom prøvene, og UPGMA som algoritme for «clustering». Total nettverkslengde av resulterende dendrogram viste forskjellen mellom prøver fra hver ekstraksjonsprosedyre. Separate analyser ble utført for hvert primersett (Bacteroidetes (HEX) og Firmicutes (FAM)) og for hver ekstraksjonsprosedyre.
Resultater
Kvantifisering av bakterielt DNA
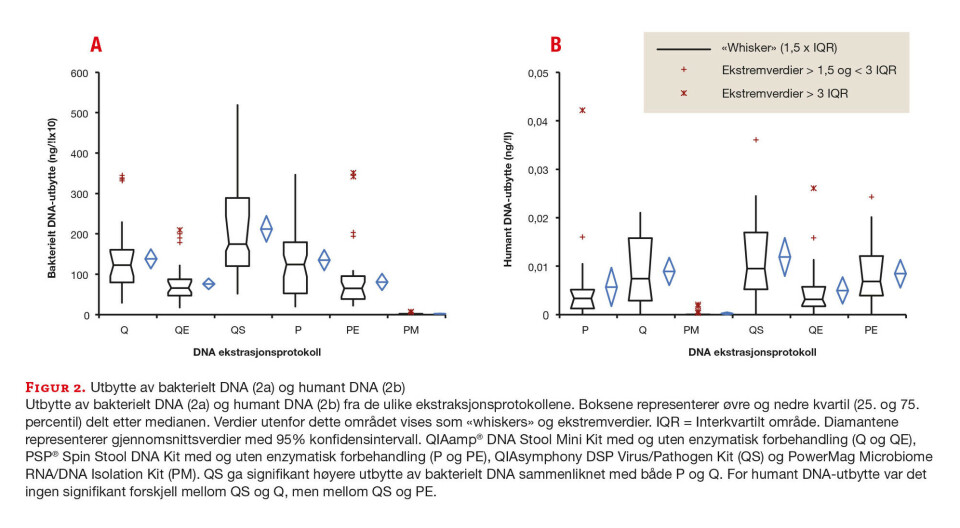
Total mengde 16S rRNA i hver prøve ble kvantifisert ved hjelp av en standardkurve med reaksjonseffektivitet på 95 % og korrelasjonskoeffisient (r2) på 0,99. ANOVA viste signifikant forskjell i bakterielt DNA-utbytte mellom protokollene (p< 0.0001). De tre protokollene med høyest bakterielt DNA-utbytte var QS, Q og P. Resultatene fra Wilcoxon rangeringstest for parvise data viste at QS ga signifikant bedre utbytte enn både P og Q (p<0.0001). Ingen signifikant forskjell ble funnet mellom P og Q (p=0.2204) (figur 2a, tabell 3).

PM ga svært dårlig utbytte av bakterielt DNA. En agarosegel-elektroforese viste at DNA fra dette kitet var degradert.
Kvantifisering av humant DNA
Total mengde humant DNA i hver prøve ble kvantifisert ved hjelp av en standardkurve med korrelasjonskoeffisient (r2) på 0.99. ANOVA viste signifikant forskjell i humant DNA-utbytte mellom protokollene (p< 0.0001). QS hadde høyest DNA-utbytte, tett fulgt av Q og PE (figur 2b). Wilcoxon rangeringstest for parvise data viste ingen signifikant forskjell mellom QS og Q (p=0.1375), men mellom QS og PE (p< 0.0001).
Bakteriell diversitet
Diversitet internt i hver prøve (alfadiversitet) varierte lite mellom de ulike ekstraksjonsprotokollene, spesielt i Firmicutes-rekken. Resultater fra Wilcoxon rangeringstest for parvise data viste imidlertid statistisk signifikant forskjell i Bacteroidetes-rekken mellom P og de to etterfølgende ekstraksjonsprotokollene (QS og Q, tabell 3).
Diversitet mellom prøver isolert med samme ekstraksjonsprotokoll viste at P ga høyest betadiversitet i Bacteroidetes-rekken, og nest høyest i Firmicutes-rekken. QS resulterte i lavest diversitet i Firmicutes-rekken og lavere diversitet enn P i Bacteroidetes-rekken (tabell 3).
Diskusjon
Målet med denne studien var å identifisere den beste DNA-ekstraksjonsmetoden for å studere bakteriell diversitet og humant DNA i fecesprøver. Fordi det er så lite humant DNA i feces, er det spesielt viktig med gode ekstraksjonsmetoder for å få best mulig utbytte. QS skilte seg ut som den protokollen som ga høyest utbytte av både bakterielt og humant DNA. P og Q har to valgfrie protokoller beskrevet av produsenten, og protokollen som er optimalisert for bakterielt DNA ble benyttet i denne studien. Forskjellen besto av et lyseringstrinn ved 95° C, i stedet for lysering ved romtemperatur. Produsentene beskriver at dette gir en høyere ratio av bakterielt/humant DNA. Dette kan ha hatt innvirkning på utbytte av humant DNA, men kan ikke forklare forskjellen i bakterielt DNA-utbytte sammenliknet med QS. QS har også tidligere vist å gi høyt utbytte av bakterielt DNA med svært god renhet når sammenliknet med andre ekstraksjonsprosedyrer (13).
Utbyttet av DNA var ikke proporsjonalt med mengde materiale inn i ekstraksjonen. Til tross for at mengde startmateriale var den samme i alle metoder, hadde protokollene ulik fortynning i løpet av ekstraksjonen. Ariefdjohan og medarbeidere viste at 10-50 mg startmateriale feces ga høyere DNA-utbytte enn startmateriale på 100-200 mg (20). Andre studier har imidlertid vist det motsatte (13). Dette illustrerer at det er viktig å gjøre egne forsøk, da man ikke nødvendigvis kan reprodusere alle parametere fra andres arbeid. PM var den protokollen med størst mengde materiale inn i ekstraksjonen, og som kom dårligst ut i vår studie. Metoden har fått gode resultater i andre studier (21) så våre data kan muligens skyldes manuell tilpasning av en protokoll som er optimalisert for et instrument.
Analyse av bakteriell diversitet illustrerte at det ikke nødvendigvis er samsvar mellom utbytte av bakterielt DNA og bakteriell diversitet. Til tross for små forskjeller i diversitet internt i hver prøve (alfadiversitet) i Firmicutes-rekken, fant vi en statistisk signifikant høyere alfadiversitet i Bacteroidetes-rekken i favør av P fremfor de andre ekstraksjonsprotokollene. Protokollen med høyest DNA-utbytte i denne studien, ga altså ikke den høyeste diversiteten. Dette er ikke i tråd med Claassen og medarbeidere som fant høyest alfadiversitet i de samme ekstraksjonsmetodene som ga størst DNA-utbytte. Årsaken til forskjellene kan være at det ble benyttet ulike prinsipper og separasjonsmetoder i diversitetsanalysene.
Ved sammenlikning av bakteriell diversitet i litteraturen er det hovedsakelig fokus på alfadiversitet, der antall bakterier og mengde av disse beregnes i ulike prøver. Vår studie har også undersøkt betadiversitet, som viser diversitet mellom prøver ekstrahert med samme protokoll. Ettersom prøvene i denne studien er fra urelaterte personer i ulike aldre, bør forskjellen mellom dem være så stor som mulig. Våre resultater tyder på at det var mindre forskjell mellom prøvene når de ble ekstrahert med QS, altså ser prøveprofilene likere ut enn for de andre ekstraksjonsprotokollene. Det kan bety at noen bakteirevarianter favoriseres og andre ikke ekstraheres like godt med QS. Forskjellene er imidlertid ikke store, og hva slags betydning eller utslag dette har i nedstrømsanalyser er usikkert. En mulig forklaring på diversitetsresultatene kan være at vi fulgte en mindre omfattende protokoll for mekanisk lysering ved ekstraksjon med QS (tabell 1). Om forskjellene i diversitet skyldes forskjellene i mekanisk lysering eller selve ekstraksjonen på instrumentet, er ikke undersøkt.
Denne studien fant ingen positiv effekt av et ekstra trinn med enzymatisk lysering. Årsaker til dette kan være at både mekanisk lysering, oppvarming og kjemisk lysering var inkludert i alle protokoller. Alle prøvene i denne studien hadde vært frosset, noe som kan ha hatt en ytterligere positiv effekt på lysering av celler og frigjøring av DNA. En annen mulig forklaring kan være at ikke alle enzymene fikk optimale forhold til å utøve sin funksjon. Lysozyme fungerer optimalt ved romtemperatur og ikke 37 ºC som benyttet i denne studien for både lysozyme, mutanolysin og lysostaphin.
Konklusjon
Ekstraksjon med QIAsymphony med DSP Virus/Pathogen kit (Qiagen) ga høyest utbytte av både humant og bakterielt DNA i denne studien. Metoden er fordelaktig med tanke på det automatiserte systemet som kan håndtere opp til 96 prøver med svært lite manuell håndtering. Ekstraksjon med PSP® Spin Stool DNA Kit (Stratec Molecular) ga høyest bakteriell diversitet. Metoden er både tid- og ressurskrevende grunnet en manuell prosedyre, men kan med fordel benyttes i bakterielle diversitetsstudier dersom tid og ressursbruk ikke er avgjørende faktorer.
Takk
Takk til Tverrfaglig Laboratoriemedisin og Medisinsk Biokjemi (TLMB), Ahus, for støtte og finansiering av arbeidet. En spesiell takk til bioingeniørene Eva Smedsrud og Gro Gundersen, Genteknologisk seksjon, TLMB, for godt samarbeid.
Interessekonflikter:
Forfatterne oppgir ingen interessekonflikter.